--samples - The file with samples names and raw data file names as explained here, can also optionally leave this out and SeekDeep will guess at what the names are based on the input data
--outDir - An output directory where analysis will be set up
--inputDir - The input raw data directory
--idFile - The id file explained above
--overlapStatusFnp - The file giving how the mates for each target overlap
Optional arguments
--lenCutOffs - A file with optional max and min lengths for the targets in the dataset
--groupMeta - A file with meta data to associate with the input samples, see above to see how this file should be associated with the other input files
--numThreads - The number of CPUs to be utilized to speed up analysis
--refSeqsDir - A directory of reference sequences to be utilized for filtering out artifacts and possible contamination, needs to have a fasta named with the name of the targets in the ID file
--replicatePatternWhenGuessing - When guessing sample names, use this to indicate replicates by giving it a regex with two regex pattern captures (captures are what’s inbetween parathesises), the first capture being the sample and the second being the replicate capture. e.g. if your input fastq are named Sample1-REP1_GAACATCG-GATCTTGC_S107_L001_R1_001.fastq.gz Sample1-REP2_GAACATCG-GATCTTGC_S107_L001_R1_001.fastq.gz you could set --replicatePatternWhenGuessing="(.*)(-REP[0-9])"
Passing on additional arguments to the default arguments of the 3 main sub-commands
Default scripts are created for each of the downstream analysis commands and additional arguments can be passed onto these scripts via the following three commands.
--extraExtractorCmds - Any extra commands to append to the default ones for the extractor step, should be given in quotes e.g. --extraExtractorCmds="--checkRevComplementForPrimers --qualWindow 50,5,18"
--extraQlusterCmds - Any extra commands to append to the default ones for the qluster step, should be given in quotes
--extraProcessClusterCmds - Any extra commands to append to the default ones for the processClusters step, should be given in quotes
overlapStatusFnp File Set up
See SeekDeep extractor and Illumina Paired Info Page for more information on overlap status input and below is a diagram of how this file should be set up and compared to the input id file.
This will extract the raw data from the input directory and it will also stitch together the mate reads, a report of how the stitching went will be in the output directory in a directory called reports. Also id files will also be copied into the directory as well. Also default scripts will be created that will run the rest of the analysis with defaults for Illumina data, all of which can be ran by the file, runAnalysis.sh in the output directory.
./runAnalysis.sh
Code
#!/usr/bin/env bash##run all parts of the pipelinenumThreads=1if[[$#-eq 1 ]];thennumThreads=$1fiSeekDeep runMultipleCommands --cmdFile extractorCmds.txt --numThreads$numThreads--rawSeekDeep runMultipleCommands --cmdFile qlusterCmds.txt --numThreads$numThreads--rawSeekDeep runMultipleCommands --cmdFile processClusterCmds.txt --numThreads$numThreads--rawSeekDeep runMultipleCommands --cmdFile genConfigCmds.txt --numThreads$numThreads--raw
The files extractorCmds.txt, qlusterCmds.txt, processClusterCmds.txt, and genConfigCmds.txt contain command line commands on each line to run the analysis. The SeekDeep runMultipleCommands is command in SeekDeep that can take in such a file and run them in parallel speeding up analysis.
See below to see how these command files match up to the pipeline.
SeekDeep Pipeline
And then to start the server to see the data interactively run the file startServerCmd.sh after running the above command files. ./startServerCmd.sh
Code
#!/usr/bin/env bash# Will automatically run the server in the background and with nohup so it will keep runningif[[$#-ne 2 ]]&&[[$#-ne 0 ]];thenecho"Illegal number of parameters, needs either 0 or 2 argument, if 2 args 1) port number to server on 2) the name to serve on"echo"Examples"echo"./startServerCmd.sh"echo"./startServerCmd.sh 9882 pcv2"exitfiif[[$#-eq 2 ]];thennohup SeekDeep popClusteringViewer --verbose--configDir$(pwd)/serverConfigs --port$1--name$2&elsenohup SeekDeep popClusteringViewer --verbose--configDir$(pwd)/serverConfigs &fi
Source Code
:::{.callout-note} # SeekDeep setupTarAmpAnalysis[back to top](#TOC)To setup the analysis use the `SeekDeep setupTarAmpAnalysis` command, see the required and optional commands below. :::```{r, engine='bash', eval=F}SeekDeep setupTarAmpAnalysis --samples sampleNames.tab.txt --outDir analysis --inputDir fastq --idFile ../idFile.tab.txt --groupMeta ../groupNames.tab.txt --lenCutOffs ../extractedRefSeqs/forSeekDeep/lenCutOffs.txt --overlapStatusFnp ../extractedRefSeqs/forSeekDeep/overlapStatuses.txt --refSeqsDir ../extractedRefSeqs/forSeekDeep/refSeqs/ ```You can also see all options by calling```{bash, eval = F}SeekDeep setupTarAmpAnalysis --help```## Required options * **\-\-samples** - The file with samples names and raw data file names as explained [here](../tutorials/tutorial_PairedEnd_noMIDs.qmd#id-file-and-samples-names-file), can also optionally leave this out and SeekDeep will guess at what the names are based on the input data * **\-\-outDir** - An output directory where analysis will be set up * **\-\-inputDir** - The input raw data directory * **\-\-idFile** - The id file explained above * **\-\-overlapStatusFnp** - The file giving how the mates for each target overlap ## Optional arguments * **\-\-lenCutOffs** - A file with optional max and min lengths for the targets in the dataset * **\-\-groupMeta** - A file with meta data to associate with the input samples, see above to see how this file should be associated with the other input files * **\-\-numThreads** - The number of CPUs to be utilized to speed up analysis * **\-\-refSeqsDir** - A directory of reference sequences to be utilized for filtering out artifacts and possible contamination, needs to have a fasta named with the name of the targets in the ID file * **\-\-replicatePatternWhenGuessing** - When guessing sample names, use this to indicate replicates by giving it a regex with two regex pattern captures (captures are what's inbetween parathesises), the first capture being the sample and the second being the replicate capture. e.g. if your input fastq are named Sample1-REP1_GAACATCG-GATCTTGC_S107_L001_R1_001.fastq.gz Sample1-REP2_GAACATCG-GATCTTGC_S107_L001_R1_001.fastq.gz you could set `--replicatePatternWhenGuessing="(.*)(-REP[0-9])"`## Passing on additional arguments to the default arguments of the 3 main sub-commands Default scripts are created for each of the downstream analysis commands and additional arguments can be passed onto these scripts via the following three commands. * **\-\-extraExtractorCmds** - Any extra commands to append to the default ones for the extractor step, should be given in quotes e.g. `--extraExtractorCmds="--checkRevComplementForPrimers --qualWindow 50,5,18"`* **\-\-extraQlusterCmds** - Any extra commands to append to the default ones for the qluster step, should be given in quotes * **\-\-extraProcessClusterCmds** - Any extra commands to append to the default ones for the processClusters step, should be given in quotes ## overlapStatusFnp File Set upSee [SeekDeep extractor](extractor_usage.html) and [Illumina Paired Info Page](illumina_paired_info.html) for more information on overlap status input and below is a diagram of how this file should be set up and compared to the input id file. 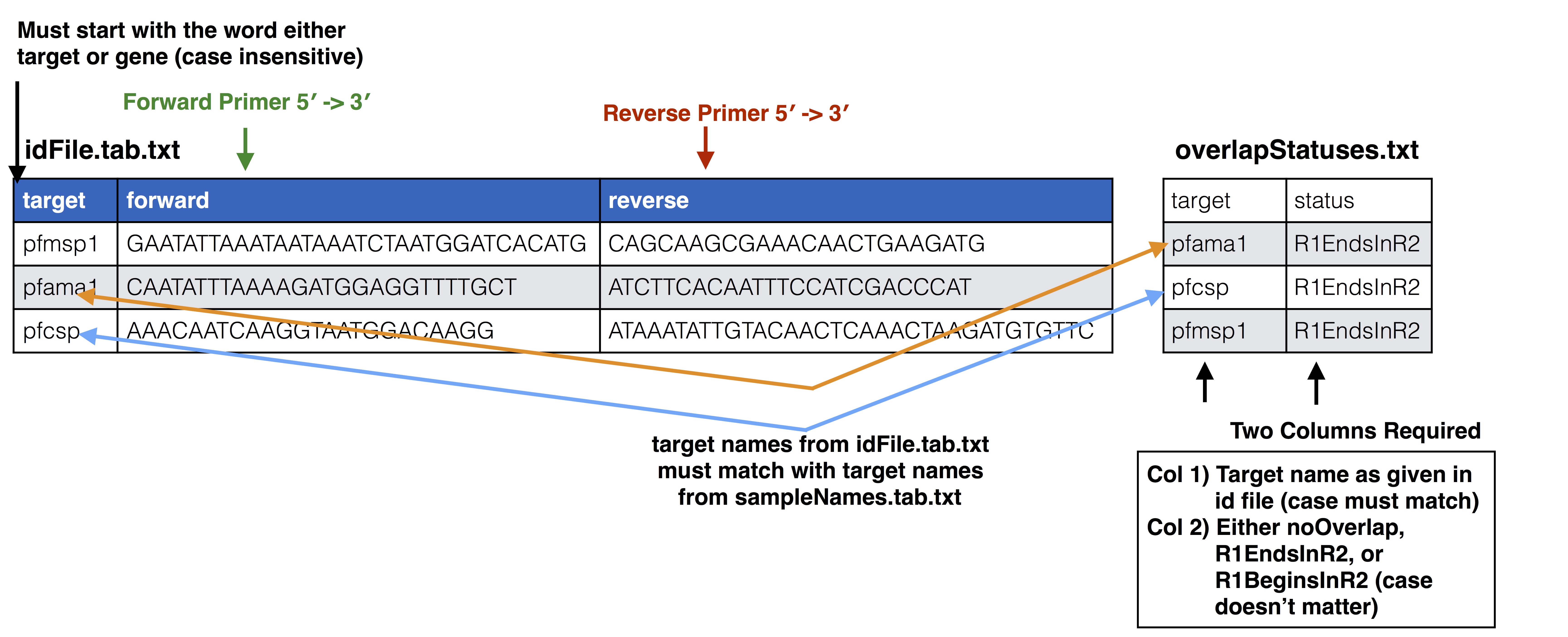This will extract the raw data from the input directory and it will also stitch together the mate reads, a report of how the stitching went will be in the output directory in a directory called reports. Also id files will also be copied into the directory as well. Also default scripts will be created that will run the rest of the analysis with defaults for Illumina data, all of which can be ran by the file, runAnalysis.sh in the output directory. **./runAnalysis.sh** ```{r, engine='bash', eval=F}#!/usr/bin/env bash##run all parts of the pipelinenumThreads=1if [[ $# -eq 1 ]]; then numThreads=$1fiSeekDeep runMultipleCommands --cmdFile extractorCmds.txt --numThreads $numThreads --rawSeekDeep runMultipleCommands --cmdFile qlusterCmds.txt --numThreads $numThreads --rawSeekDeep runMultipleCommands --cmdFile processClusterCmds.txt --numThreads $numThreads --rawSeekDeep runMultipleCommands --cmdFile genConfigCmds.txt --numThreads $numThreads --raw```The files extractorCmds.txt, qlusterCmds.txt, processClusterCmds.txt, and genConfigCmds.txt contain command line commands on each line to run the analysis. The `SeekDeep runMultipleCommands` is command in `SeekDeep` that can take in such a file and run them in parallel speeding up analysis.See below to see how these command files match up to the pipeline. 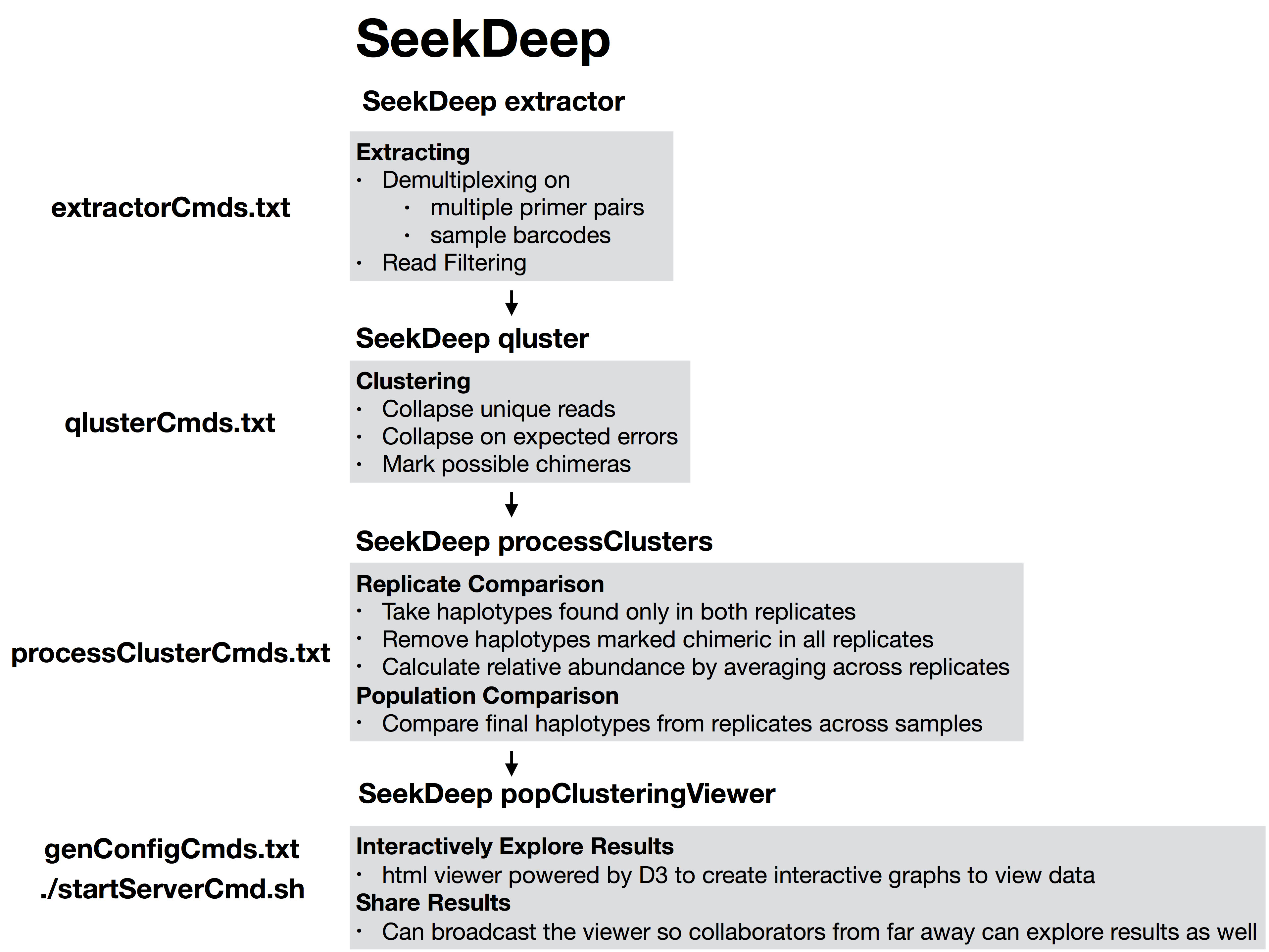And then to start the server to see the data interactively run the file `startServerCmd.sh` after running the above command files. **./startServerCmd.sh** ```{r, engine='bash', eval=F}#!/usr/bin/env bash# Will automatically run the server in the background and with nohup so it will keep runningif [[ $# -ne 2 ]] && [[ $# -ne 0 ]]; then echo "Illegal number of parameters, needs either 0 or 2 argument, if 2 args 1) port number to server on 2) the name to serve on" echo "Examples" echo "./startServerCmd.sh" echo "./startServerCmd.sh 9882 pcv2" exit fiif [[ $# -eq 2 ]]; then nohup SeekDeep popClusteringViewer --verbose --configDir $(pwd)/serverConfigs --port $1 --name $2 &else nohup SeekDeep popClusteringViewer --verbose --configDir $(pwd)/serverConfigs & fi```

