Code
SeekDeep extractor --help
SeekDeep extractorPairedEnd --help The main purpose of extractor is to take raw data and demultiplex by sample barcodes if present and by primer pairs. It at most requires two arguments: an input file which can be fastq (--fastq) or fasta (--fasta) and an ID file (--id) that supplies primer pairs and barcodes if present. It will also do some quality control as well for length and quality scores.
There is a separate extractor command for extracting paired end data and is simply called SeekDeep extractorPairedEnd and shares most of the command line options with regular single end SeekDeep extractor
Calling -help will give a help message on running the program
SeekDeep extractor --help
SeekDeep extractorPairedEnd --help Also all flags in SeekDeep are case insensitive and so all the following would have the same results
SeekDeep extractor --help
SeekDeep extractor --HELP
SeekDeep extractor --HeLP
SeekDeep extractor --HeLp
SeekDeep extrActor --HeLP
SeekDeep ExtraCtor --HeLp
SeekDeep EXTRACTOR --HeLpThe id file is a tab delimited file that contains both primer pair info and optional barcode information
Primer information starts with a header line that must start with either gene or target and have two additional columns for forward and reverse primers, the line can start with either gene or target.
target forward reverse
PFAMA1 CAGGGAAATGTCCAGTATT CTTGAACATAAAGTCAATTC
PFCSP ACAATCAAGGTAATGGACAAGG TTTTCAATATCATTTYCATAATCTAATTThe primers can contain ambigious bases (Y,R,etc.) and should be all upper case
Forward primer should be in the 5`-3` direction.
The reverse primer should in in the 5`-3` direction as well.
If data is multiplexed the MID name and corresponding barcode information comes after the primer information. Barcode information needs to start with a header id barcode which the id is the important name for parsing. Mids should be in the direction they would be found in if reads are found in the direction of the forward primer. It is always assumed that the mid and primer set up is MID-PRIMER. If this is not the case for your data contact Nick Hathaway (nicholas.hathaway@umassmed.edu) to see if he can add options for accommodating your data structure.
target forward reverse
PFAMA1 CAGGGAAATGTCCAGTATT CTTGAACATAAAGTCAATTC
PFCSP ACAATCAAGGTAATGGACAAGG TTTTCAATATCATTTYCATAATCTAATT
id barcode
MID01 ACGAGTGCGT
MID02 ACGCTCGACA
MID03 AGACGCACTC
MID04 AGCACTGTAG
MID05 ATCAGACACG
MID06 ATATCGCGAG
MID07 CGTGTCTCTA
MID08 CTCGCGTGTC
MID10 TCTCTATGCG
MID11 TGATACGTCT
MID13 CATAGTAGTG
MID14 CGAGAGATAC
MID15 ATACGACGTA
MID16 TCACGTACTA
MID17 CGTCTAGTAC
MID18 TCTACGTAGCA directory will be created for the output of extractor, this defaults to the name of the file plus the work extractor and then the current date and time of the when the command was run. This can be changed with the --dout flag. In this directory will be the sequences and several files reporting on how the extraction when.
Example of extractionProfile.tab.txt
cat: extraFiles/exampleExtraction/extractionProfile.tab.txt: No such file or directoryExample of extractionStats.tab.txt
cat: extraFiles/exampleExtraction/extractionStats.tab.txt: No such file or directoryThe output files will be named by the using the name given in the gene column and the name in id column if the data is multiplexed.
ls ./PFAMA1MID01.fastq
PFAMA1MID02.fastq
PFAMA1MID03.fastq
PFAMA1MID04.fastq
PFAMA1MID05.fastq
PFAMA1MID06.fastq
PFAMA1MID07.fastq
PFAMA1MID08.fastq
extractionProfile.tab.txt
extractionStats.tab.txt
parametersUsed.txt
runLog_extractor.txtSeekDeep extractor and SeekDeep extractorPairedEnd require an input sequence file and an identifier file.
SeekDeep extractor --fastq example.fastq --id ids.txt
SeekDeep extractor --fastqgz example.fastq.gz --id ids.txtSeekDeep extractor --fasta example.fasta --id ids.txtSeekDeep extractor --fasta example.fasta --qual example.fasta.qual --id ids.txt
#or
SeekDeep extractor --stub example --id ids.txtFor paired end the only options are fastq or fastqgz and both mates have to be indicated. This also assumes that R1 sequences are in the 5`-3` direction of the top strand, and R2 are in the 5`-3` direction of the bottom strand.
SeekDeep extractorPairedEnd --fastq1 example_R1.fastq --fastq2 example_R2.fastq --id ids.txt
SeekDeep extractorPairedEnd --fastq1gz example_R1.fastq.gz --fastq2gz example_R2.fastq.gz --id ids.txtMultiplexing simply requires having MID/barcodes in id file, a header line is required that starts with id to indicate to SeekDeep extractor that barcodes are starting.
cat id_file.tab.txttarget forwardPrimer reversePrimer
PFAMA1 CCATCAGGGAAATGTCCAGT TTTCCTGCATGTCTTGAACA
id barcode
MID01 ACGAGTGCGT
MID02 ACGCTCGACA
MID03 AGACGCACTC
MID04 AGCACTGTAG
MID05 ATCAGACACG
MID06 ATATCGCGAG
MID07 CGTGTCTCTA
MID08 CTCGCGTGTCSeekDeep extractor --fastq example.fastq --id id_file.tab.txt --multiplexMultiple primer pairs is done simply by adding another primer info line, this can also be combined with multiplexing
cat multipleGenePairs.id.txtgene forwardPrimer reversePrimer
PFAMA1 CCATCAGGGAAATGTCCAGT TTTCCTGCATGTCTTGAACA
PFMSP1 AACTAGAAGCTTTAGAAGATGCA ACATATGATTGGTTAAATCAAAG
id barcode
MID01 ACGAGTGCGT
MID02 ACGCTCGACA
MID03 AGACGCACTC
MID04 AGCACTGTAG
MID05 ATCAGACACG
MID06 ATATCGCGAG
MID07 CGTGTCTCTA
MID08 CTCGCGTGTCSeekDeep extractor --fasta example.fasta --id multipleGenePairs.id.txt For paired reads an additional file is required that indicates if there is overlap between the mates. There are three possible overlap scenarios: noOverLap, r1EndsInR2 (normal overlap goal), and r1BeginsInR2 (happens with read through). If there is overlap SeekDeep extractorPairedEnd will stitch together the mates and if there is no overlap it will extract and have a _R1 and _R2 files for the target. The different overlap scenarios are required as there are many ways to generate artifacts in PCR (unspecific amplification, primer dimers, etc) that could lead to the wrong overlap and therefore reads that overlap in the way that they are expected to are given in the final output files and the rest assumed to be artifact. See Illumina Paired Info Page for a diagram on how sequences overlap.
The overlap status of the targets is given by a file with two columns, target and status. The order of columns does not matter and the case of the status column does not matter, the target column does. The name given in the target column must match with the names given in the id file.
cat overlapStatuses.txttarget status
PFAMA1 R1BeginsInR2
PFCSP R1BeginsInR2SeekDeep extractor --fastq1 example_R1.fastq --fastq2 example_R2.fastq --id multipleGenePairs.id.txt --overlapStatusFnp overlapStatuses.txt Reads are filtered on expected length by choosing a minimum length (--minLen) and a maximum length (--maxLen), these default to be within 20% of the median input read lengths
SeekDeep extractor --fastq example.fastq --id ids.txt --minLen 150SeekDeep extractor --fastq example.fastq --id ids.txt --minLen 150 --maxLen 300You can also supply a file with len cutoffs which is especially useful for when you have different expected lengths for different targets, the file needs to contain at least three columns with one column named target which contain the name of the target region which must match the target name in the --id file and two columns minlen and maxlen which contain the min and max length for the target.
This is supplied with the --lenCutOffs flags.
SeekDeep extractor --fastq example.fastq --id ids.txt --lenCutOffs lengthCutOffsForTargets.tab.txt cat lengthCutOffsForTargets.tab.txttarget minlen maxlen
PFAMA1 200 240
PFCSP 254 294When quality values are supplies they can be used to filter reads as well, there are two options for quality filtering. The default is to use the sliding quality window threshold method, this is good for Ion Torrent, 454, and PacBio. The second quality option is good for Illumina
By default a sliding window of size 50 and stepping by every 5 bases and checking to see if the average quality of those 50 bases are above a threshold of 25. The default values can be changed by using the --qualWindow flag and supplying three values separated by commas which are the window size, window step, and window threshold. So the below example would use a window size of 50, a step of 5, and a average threshold of 20
SeekDeep extractor --fastq example.fastq --id ids.txt --qualWindow 50,5,20Also the default behavior is to throw out any read where this ever fails and this can be changed to trimming the read at the failed window instead using the -qualWindowTrim flag. The minimum length filter is applied after this step so if the trim falls below the min len it will still be filtered out
SeekDeep extractor --fastq example.fastq --id ids.txt --qualWindow 50,5,20 --qualWindowTrimA second option for filtering using quality scores is using the distribution of a reads quality scores. This is done by choosing a quality score cut off and checking to see if a certain fraction of quality scores are above a fraction cut off. This is done by using the -qualCheck and the qualCheckCutOff flags. So the below example would throw out any reads where less than 75% of their quality scores was above 25
SeekDeep extractor --fastq example.fastq --id ids.txt --qualCheck 25 --qualCheckCutOff .75Since extraction might take some time, it might be nice to see how many reads you would loose with different setting, so a command was added to SeekDeep to do a dry run to output the percentage of reads you would loose if certain quality parameters were used, this is called SeekDeep dryRunQualityFiltering and the arguments are similar to the arguments above.
SeekDeep dryRunQualityFiltering --fastq example.fastq --qualWindow 50,5,25void njhseq::SeqInput::openInLockFree(): Error file: [31m[1mMultiplex_IT_Tutorial_Materials/IonTorrent1.fastq[0m doesn't existSeekDeep dryRunQualityFiltering --fastq example.fastq --qualWindow 50,5,20void njhseq::SeqInput::openInLockFree(): Error file: [31m[1mMultiplex_IT_Tutorial_Materials/IonTorrent1.fastq[0m doesn't existSeekDeep dryRunQualityFiltering --fastq example.fastq --qualWindow 50,5,18void njhseq::SeqInput::openInLockFree(): Error file: [31m[1mMultiplex_IT_Tutorial_Materials/IonTorrent1.fastq[0m doesn't existSeekDeep extractor --fastq example.fastq --id ids.txt --qualCheck 25 --qualCheckCutOff .75void njhseq::SeqInput::openInLockFree(): Error file: [31m[1mMultiplex_IT_Tutorial_Materials/IonTorrent1.fastq[0m doesn't existSeekDeep extractor --fastq example.fastq --id ids.txt --qualCheck 20 --qualCheckCutOff .75void njhseq::SeqInput::openInLockFree(): Error file: [31m[1mMultiplex_IT_Tutorial_Materials/IonTorrent1.fastq[0m doesn't existThere are various filtering parameters that can be applied to the presence of primers. This includes percent of primer found, the number of mismatches to the primer, and not searching for the primer at all. See SeekDeep extractor -help for more details and defaults
Looking for primers can be turned off and simple filtering can be done, this requires the flag --noPrimers and that the ID file has just one primer line so that a target name can be given to the out files but the forward and reverse primer columns are ignore.
SeekDeep extractor --fastq example.fastq --id ids.txt --noPrimers Depending on the library set up reads are can be found in two directions. Reads found in the reverse complement direction will be reverse complemented and their name will be marked with _Comp so all sequences are in the same direction. There are separate flags for searching MIDs in both directions (–checkRevComplementForMids) and for searching for primers in the both directions (–checkRevComplementForPrimers). And depending on library prep again you might have to combine the two.
SeekDeep extractor --fasta example.fasta --id ids.txt --checkRevComplementForMidsSeekDeep extractor --fasta example.fasta --id ids.txt --checkRevComplementForPrimersSeekDeep extractor --fasta example.fasta --id ids.txt --checkRevComplementForMids --checkRevComplementForPrimersBarcodes and primers aren’t always located at the very beginning of the sequences and the search region has to be expanded which can be done using the --midWithinStart flag for mids and --primerWithinStart, the below example would check for the barcodes and primers for start sites within the first 10 bases of the start of the sequence (the primer/MID just has to start within this many bases and not be contained completely with this region, e.g. the primer could be found at base 10 but extend to base 30 and would be caught with this flag). When data is multiplexed the primer within start parameter is the number of bases within the found MID and not the start of the sequence.
SeekDeep extractor --fastq example.fastq --id ids.txt --midWithinStart 10SeekDeep extractor --fastq example.fastq --id ids.txt --primerWithinStart 10And again, depending on library prep you might need to combine these two flags. For instance, if you had say a variable number of bases of up to 4 in front of your MIDs and then an additional variable number of bases in between your MIDs and primers (lets say up to 10).
SeekDeep extractor --fastq example.fastq --id ids.txt --midWithinStart 4 --primerWithinStart 10It is advised to keep these numbers low as a high number will greatly increase the search space and could lead to many false positives.
Dual barcoding schemes can be done by adding another column to the barcode portion of the input ID file. The first column is assumed to be associated with the forward target primer and the second column is assumed to be associated with the
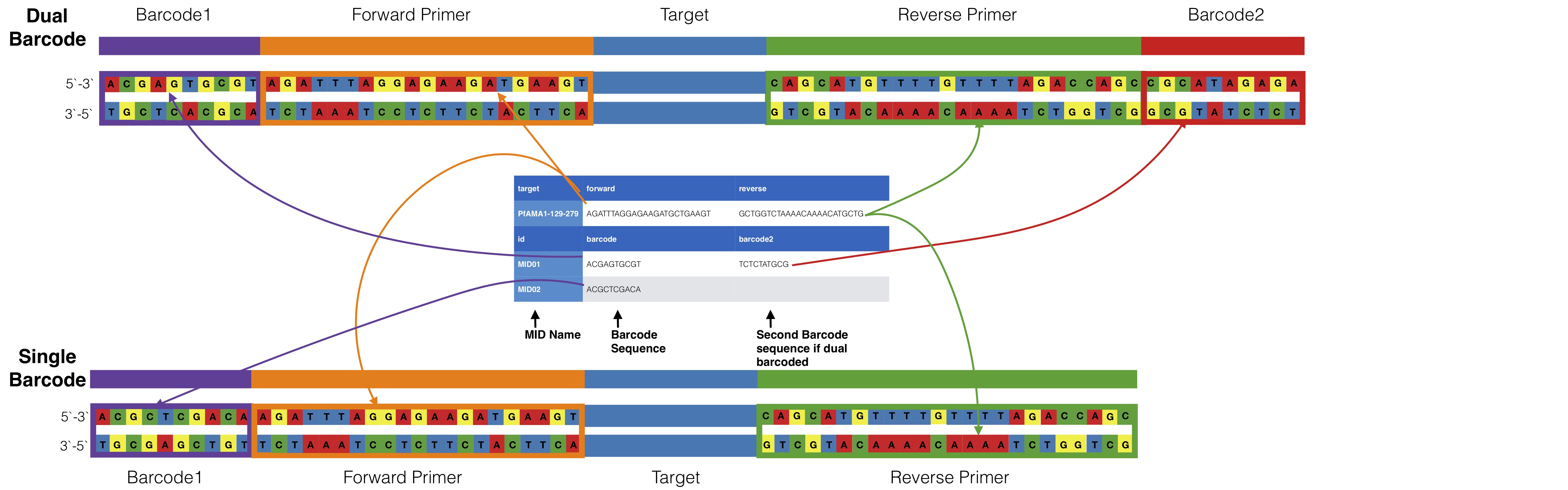
SeekDeep extractor --fastq example.fastq --id ids.txtYou can allow mismatches in barcodes using --barcodeErrors
SeekDeep extractor --fastq example.fastq --id ids.txt --barcodeErrors 2The names given to the sequences are sometimes annoying so adding the --rename flag will rename the sequence with what primer/mid they were found with
SeekDeep extractor --fastq example.fastq --id ids.txt --renameA file will be created called renameKey.tab.txt which contains the name conversion.
cat renameKey.tab.txtoriginalName newName
JUW96:00012:00129 PFAMA1MID01.00
JUW96:00013:00133 PFAMA1MID01.01
JUW96:00017:00147 PFAMA1MID01.02_Comp
JUW96:00018:00120 PFAMA1MID01.03
JUW96:00020:00136 PFAMA1MID01.04_Comp
JUW96:00029:00127 PFAMA1MID01.05
JUW96:00032:00129 PFAMA1MID01.06
JUW96:00033:00147 PFAMA1MID01.07_Comp
JUW96:00035:00137 PFAMA1MID01.08_CompTo change the default directory name use the --dout flag. SeekDeep will never overwrite a directory if it already exists and will fail and quit if it tries to create a directory that exists.
SeekDeep extractor --fastq example.fastq --id ids.txt --dout extractionDirThe dout option also understand the key work TODAY to mean to insert the current date and time there instead though this means a output directory name can never have TODAY all in caps in it
SeekDeep extractor --fastq example.fastq --id ids.txt --dout extractionDir_TODAY